
The Hallmarks of Cancer are the ten characteristics that differentiate a cancer cell from a normal cell. Over the course of a year, science translator Dr. Buddhini Samarasinghe tackled each of the the ten Hallmarks of Cancer in a guest blog series for Scientific American.
The blogs break down the basic biology of each Hallmark and what happens when the system breaks down. It is important to understand these Hallmarks as we investigate the influence of chemicals and how they may act individually and in groups to disrupt enough of these Hallmark mechanisms to cause cancer.
Currently nine out of the ten blogs have been published, and the tenth is being prepared.
In 2000, Robert Weinberg and Douglas Hanahan published a review article in the journal Cell titled "The Hallmarks of Cancer". It was a seminal paper in every sense of the word; downloaded 20,000 times a year between 2004 and 2007, with over 15,000 citations in other research papers. In 2011, Weinberg and Hanahan updated their list by proposing four more new Hallmarks of Cancer, bringing up the list to Ten Hallmarks of Cancer.
Why is this paper so important? Cancer, as we know by now, is an incredibly complicated disease. A single tumor sample could have over a hundred different mutations; nearly one in every two hundred genes in the human genome. If two breast cancer specimens are compared, the set of mutated genes are far from identical. Every tumor is unique. Weinberg and Hanahan simplified this dauntingly complex disease to six underlying principles. The hugely complicated beast that is cancer, so diverse that even the same organ can have many different tumor types, was reduced to just six common traits that every single cancer shares, to facilitate that transformation from a normal cell to a cancer cell. It answers the 'how does cancer happen' question very elegantly, and we gain insight into all the different things that go wrong in a cancer cell.
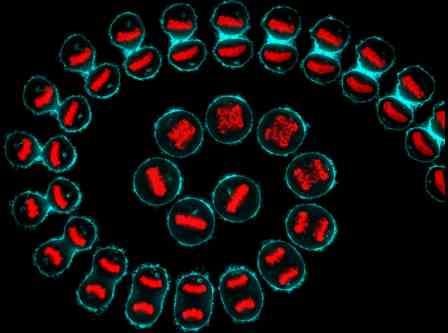 A composite from a time-lapse of a hela cell (cervical cancer) undergoing cell division. Cellular structures have been visualized using cyan (cell membrane) and red (DNA). Image credit: Kuan-Chung Su, London Research Institute, Cancer Research UK, Wellcome Images.
A composite from a time-lapse of a hela cell (cervical cancer) undergoing cell division. Cellular structures have been visualized using cyan (cell membrane) and red (DNA). Image credit: Kuan-Chung Su, London Research Institute, Cancer Research UK, Wellcome Images.However, the science is not accessible to the public because the molecular mechanisms described require a specialist knowledge in the field of cell biology. I will go through each of these Hallmarks in detail and will explain the normal processes that occur inside the cell and then explain what goes wrong with this process in a cancer cell.
Cancer is so prevalent, and is a topic that we hear about on a daily basis. Cancer is also deeply personal; we know people who are directly affected by cancer, either themselves or a loved one. Not everyone has the knowledge to understand what is going on, and it can be very scary to hear the jargon from a doctor or an oncologist. To many people, cancer is the big scary 'C-word'. Trying to research it online is not usually helpful, as even Wikipedia has a confusingly large amount of jargon. This series will address that knowledge gap. It will demystify cancer, and answer the question 'why does cancer happen?'
Reprinted with permission of Buddhini Samarasinghe and Scientific American, September 11, 2013
1. Self-Sufficiency in Growth Signals
The Hallmarks of Cancer are ten anti-cancer defense mechanisms that are hardwired into our cells, that must be breached by a cell on the path towards cancer. The first Hallmark of Cancer is defined as "Self-Sufficiency in Growth Signals". What does this mean? Before I explain how growth signals are intimately involved in the development of cancer, it is necessary to define and understand what growth factors are, and explain how they control normal cellular behavior.
Growth Factors
Growth factors are, simply put, substances that control the multiplication of cells. There are many different types of growth factors, but they all have several characteristics in common. They are all proteins, and present at very low concentrations in tissues but with a high biological activity. They are responsible for controlling essential functions within the cell; growth, specialization and survival. Growth factors also do not circulate in the blood stream; instead, they act locally in areas near the cells that produce them. The image below shows a growth factor known as Vascular Endothelial Growth Factor (VEGF).
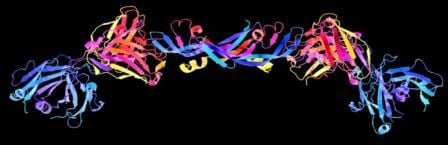 Growth factors are beautiful! This is a 3D schematic representation (also known as a ribbon diagram) of the structure of a growth factor known as vascular endothelial growth factor (VEGF). VEGF stimulates the development of new blood vessels, a process known as angiogenesis. Many large tumors secrete their own supply of VEGF in order to generate a supply line of oxygen-rich blood for the growing tumor to feed on. Image credit: Gizmag
Growth factors are beautiful! This is a 3D schematic representation (also known as a ribbon diagram) of the structure of a growth factor known as vascular endothelial growth factor (VEGF). VEGF stimulates the development of new blood vessels, a process known as angiogenesis. Many large tumors secrete their own supply of VEGF in order to generate a supply line of oxygen-rich blood for the growing tumor to feed on. Image credit: Gizmag
Cell Signaling
It is impossible to talk about growth factors and cancer without going over some of the basics of cell signaling. We are multi-cellular animals, and as such, our cells need to communicate with each other, so they can act in a coordinated manner in response to the environment. The basis of this communication comes from a process known as cell signaling.
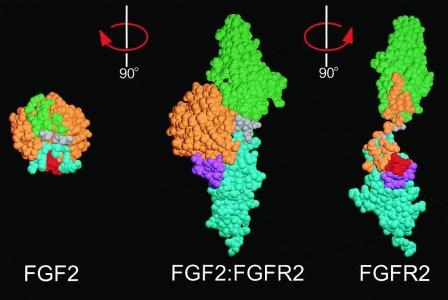
Growth factors fit perfectly into growth factor receptor binding sites. Two different types of fibroblast growth factor (FGF1 and FGF2, left) shown bound to its specific receptor (center) and separate (right). Image credit: Alexander Plotnikov.
The behavior of a cell depends on its immediate surrounding environment, known as the microenvironment. The assortment of growth factors in this microenvironment is the most important aspect regulating the behavior of that cell. All growth factors exert their effects by binding to a receptor. Receptors are proteins found on the surface of a cell that receive such chemical signals from the outside of the cell. Each growth factor has it's own receptor; think of it as a key (the growth factor) fitting into a lock (the receptor). Growth factor receptors tend to be 'transmembrane molecules'; this means that one end of the receptor 'sticks out' through the cell membrane into the microenvironment while the other end projects inside the cell. By spanning across the cell membrane, growth factor receptors are able to communicate signals from outside the cell (e.g. presence of growth factors in the microenvironment) to the inside of the cell. Revisiting the lock and key analogy, think of it as a key that fits into a lock that protrudes through the door-frame, instead of being flush against the door.
The binding of the growth factor to its specific receptor triggers a phosphorylation reaction inside the cell. Phosphorylation, or the addition of a phosphate group to a protein molecule, is an important step in cell signaling. This is because many proteins exist in an 'on' or 'off' state that can be switched by phosphorylation. Therefore, phosphorylation is a key step in regulating their activity. The enzymes that add phosphate groups to proteins are known as kinases; enzymes that remove phosphates are known as phosphatases. The exterior end of the receptor protein (the bit that sticks out of the cell) carries the growth factor binding site; the other end which projects inside the cell carries a kinase site. Binding of growth factor to the receptor binding site activates the kinase domain on the interior end of the receptor protein. This activated kinase, true to it's name, then goes on to add phosphate groups to other proteins inside the cell, which then activate more proteins downstream, triggering a signaling cascade that finally ends with the activation of genes that bring about... you guessed it, cellular growth, specialization, or survival! The image below illustrates this process - I couldn't find a decent one online so I made my own!
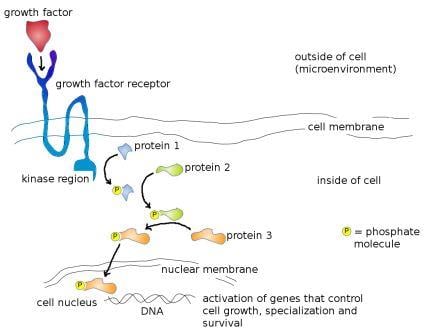
Mode of action of a typical growth factor. growth factor (red) binds to specific growth factor receptor binding site (dark blue) on cell surface, which activates the kinase region (light blue). Activated kinase region now adds a phosphate group (yellow) to protein 1 (blue) which activates it. Activated protein 1 now adds a phosphate group to protein 2 (green) further down the pathway, which activates it. Activated protein 2 subsequently adds a phosphate group to protein 3 (orange) which activates it. Activated protein 3 moves through the nuclear membrane into the cell nucleus where it physically binds to the DNA and activates genes that control cell growth, specialization and survival. Image credit: Buddhini Samarasinghe
The description above is an extremely simplified version of what happens inside a cell; in reality, it is not so much a linear signaling pathway as it is an interwoven, intricate signaling web, with promiscuous proteins from many different pathways activating and repressing one another. The image below is not meant to frighten you (!) but rather to give you an idea how truly complex just one such signaling pathway, known as the MAPK/Erk pathway is.
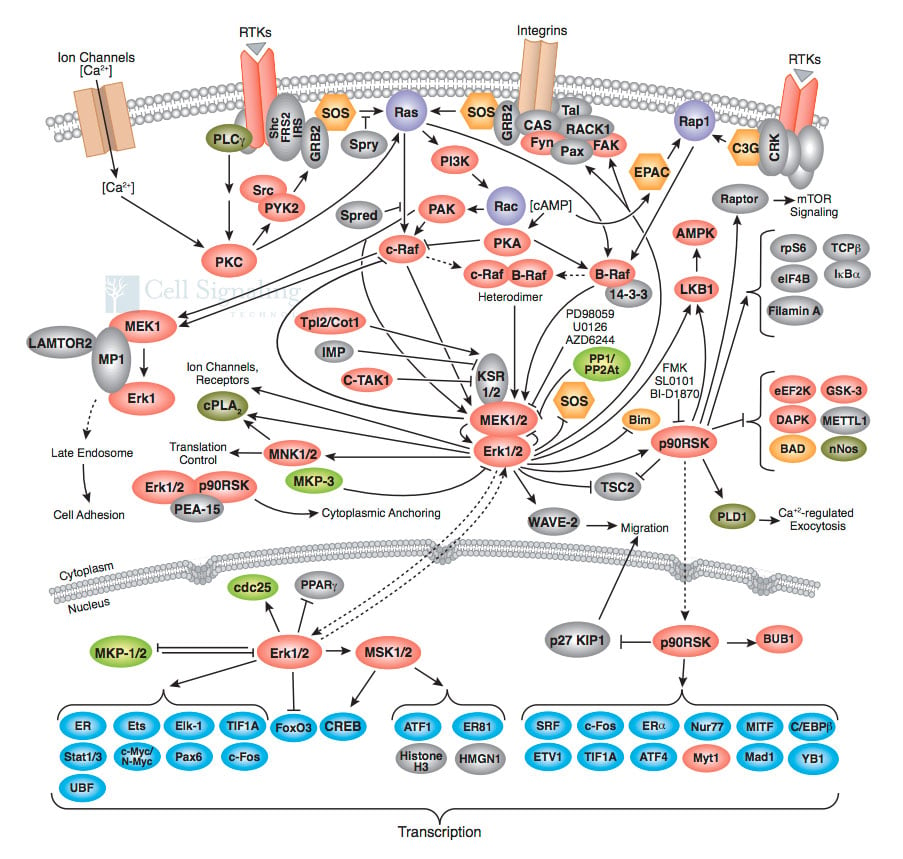
A truly complex web of cell communication! These are some of the proteins we know that are involved in a single pathway known as the MAPK/Erk pathway. Signals from the outside of the cell go through this web of signaling, ultimately ending up with the activation of genes involved in growth, specialization and survival of the cell. Image credit: Cell Signaling Technology.
Becoming Independent
So now that we understand the basics of the molecular mechanisms behind cell signaling, what happens in a cancer cell that turns this orderly process so horribly awry? As you may have figured out, normal cells cannot divide without the 'go ahead' from growth factors. Even normal cells growing on a petri dish need growth factors supplied from animal serum to divide; if not they enter a dormant state and eventually die. Cancer cells on the other hand, do not need this 'go ahead'. This liberation from being dependent on externally supplied growth factors removes a very critical checkpoint on the path towards cancer.
Breaching the Defenses
How do cancer cells bypass this checkpoint? There are three common strategies:
First, they can alter the level of growth signal itself. Normally, growth factors are made by one type of cell in order to act on another type of cell. However many cancer cells acquire the ability to synthesize and secrete their own growth factors, stimulating others of their kind, which creates a feedback loop in which more cancer cells divide under the influence of growth factor to synthesize more growth factor and so on. If the key to the lock is typically provided by a caretaker, then this means having your own DIY key-cutting machine, so that dependence on a locksmith is eliminated.
Second, the cancer cell can tweak the growth factor receptor itself, so that a larger-than-normal number of these receptors are present on the surface of the cancer cell. This means that the cancer cell becomes hyper-responsive to ambient levels of growth factor that would normally not be enough to trigger cell division. Additionally in some tumors, the growth factor receptor is structurally altered, sometimes lacking the regulatory regions, which results in the switch being permanently stuck at 'on'. There is no need for a key, the lock opens without one.
Finally, there are alterations further downstream of the signaling pathway, so that the requirement for growth factor and receptor are both bypassed. For example, one of the key downstream components of the growth factor signaling pathway is a protein known as Ras. Mutant Ras is permanently 'switched on'. Mutant Ras is the most common gene in human cancer; 25% of all human tumors, and up to 90% of certain types of cancer such as pancreatic cancer have mutant Ras. Why bother with trying to unlock a door if the walls don't exist?
It is worth remembering that cancer cells cannot do what they do in isolation. The apparently normal bystanders, such as cells of the nearby blood vessels and connective tissue must also play key roles in driving cancer cell growth. In normal tissues, cells are instructed to grow by their neighbors; this is true of the tumor microenvironment as well. A tumor is not only made of cancer cells. Tumors should be regarded as complex tissues in which the mutant cancer cells have co-opted and subverted normal neighbor cells by inducing them to release growth factors as well.
With these three strategies for achieving self-sufficiency in growth signals, cancer cells can successfully breach one of the ten anti-cancer defenses hardwired into our normal cells. The result is cells that are capable of growing uncontrollably, unstoppably and pathologically - in other words, cancer cells.
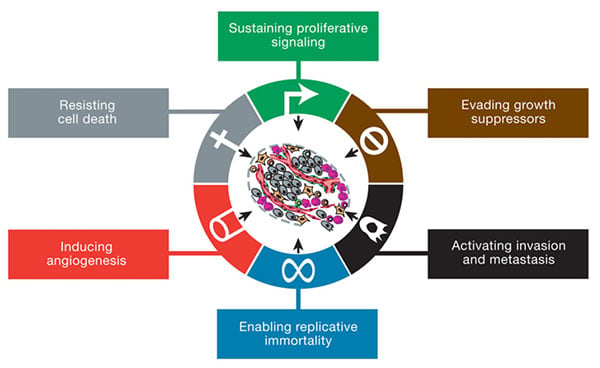
The six hallmarks of cancer, as published in 2000 by Weinberg and Hanahan. Image credit: Weinberg and Hanahan.
Fighting back
How can we use this knowledge about cancer to fight back? This is where the magic words 'targeted therapies' come in. Since cancer cells hijack normal growth factor response pathways to become self sufficient, it is logical for us to target these errant pathways specifically. If a growth factor receptor is stuck on 'always on', for example as a perpetually active kinase, then finding a specific inhibitor to stop the activity of this kinase would starve the cancer cell of the signal it is dependent on for uncontrolled growth.
Two such drugs, discovered in the 1990s utilize this principle. Gleevec, used as treatment for chronic myelogenous leukemia and Herceptin, for the treatment of breast cancer, both inhibit specific components of growth factor response pathways to starve the cancer of this signal. Earlier in this article I mentioned the Ras protein, which is frequently mutated in cancers; work is currently underway to find small molecules that are capable of inhibiting Ras. An exciting era of targeted cancer therapies lie ahead of us, because we have a deeper understanding of how cancer happens.
Reprinted with permission of Buddhini Samarasinghe and Scientific American, September 18, 2013
2. Insensitivity to Antigrowth Signals
Before I explain how failure to respond to antigrowth signals is closely involved in the development of cancer, it is useful to define and understand how cell division works. Cancer is, after all, the uncontrolled division of the cell; so we first need to understand how normal cell division is controlled through the cell cycle.
The Cell Cycle
Think of the Cell Cycle as the control system of a washing machine. A washing machine passes through several stages in a wash cycle; soaking the clothes, adding detergent at the correct time, rinsing the clothes for the appropriate duration to remove the detergent, adding the fabric softener at the correct time, a final rinse and then spinning the clothes to remove as much water as possible. In much the same way, the cell cycle is a series of tightly regulated events inside a cell that lead to its division into two daughter cells. In between these start and end states, the DNA inside the parent cell needs to double and then be divided equally between the two daughter cells. Intricate feedback loops of responsive proteins trigger events in the cell cycle, guiding the cell through checkpoints that lie between every stage. These checkpoints are important because they act as safety valves, ensuring that an errant, incorrectly dividing cell with damaged DNA is promptly whisked away and destroyed rather than allowed to continue its life.
The Four Stages
The cell cycle has four stages.
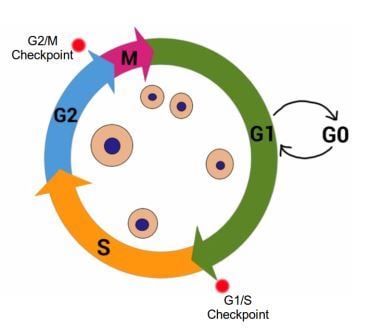
The cell cycle, showing G1, A, G2 and M stages. cell growth occurs during G1 and G2, while DNA is synthesized in S stage. Cell division occurs during M stage (mitosis). G0 indicates quiescent stage where the cell exits the cell cycle but can re-enter if the signals from the environment are appropriate. Red dots indicate important cell cycle checkpoints G1/S and G2/M. Image credit: Buddhini Samarasinghe
Gap 1 (G 1) - during this stage the cell is actively growing in size, and preparing the required components for DNA synthesis. During and at the end of this stage, the cell monitors its surrounding environment (the microenvironment) to make sure there aren't any 'stop!!' signals. This is known as the G1/S Checkpoint.
Synthesis (S) - DNA replication takes place during this stage. If there is damaged DNA in the cell, it should not be replicated, and the G1/S checkpoint ensures this. The G1/S Checkpoint is extremely important to prevent the replication of damaged DNA.
Gap 2 (G2) - The cell continues to grow in the G2 stage following DNA replication. At this stage, it is vital to recognize any damaged DNA before proceeding with cell division. This constitutes the G2/M Checkpoint. If the cell detects damage to its DNA, it will not proceed to the fourth and final stage of the cell cycle. The first G1/S Checkpoint ensures that the template DNA prior to replication is undamaged, and the G2/M Checkpoint ensures that the newly replicated DNA is error-free before proceeding with cell division.
Mitosis (M) - This stage represents the culmination of the cell cycle. Cell growth stops and the cell divides into two equal daughter cells. Some cells enter a G0 stage known as quiescence which can be considered a place of rest. A cell in G0 has exited the cell cycle, and is neither dividing nor preparing to divide; however, the cell is still alive and actively metabolizing, it has simply stopped dividing. The cell may re-emerge from this quiescent stage back into the cell cycle, given the right signals from its microenvironment. Some cells permanently exit the cell cycle, moving to a post-mitotic state. There is no coming back from this path; it is usually associated with mature cells that have differentiated, i.e. taken their adult form. Therefore a cell, at the basic level, has three choices facing it: continue to grow and divide by staying in the cell cycle, take a temporary break by entering G0, or permanently exit the cell cycle into the post-mitotic state.
Controlling the Cell Cycle: Cyclins and CDKs
Two key classes of regulatory proteins control the checkpoints within the cell cycle. These proteins are known as Cyclins and Cyclin Dependent Kinases (CDKs). Cyclins and CDKs cannot work without each other; they need to 'team up'. Cyclins provide the regulation for the team, and CDKs are the catalyst. CDKs are active only with their specific partner cyclins; this is why they are known as cyclin dependent kinases. In the previous Hallmark of Cancer article, I explained that many proteins exist in an 'active' or 'inactive' state that can be switched by phosphorylation (i.e. adding a phosphate group to the protein), and that kinases are enzymes that add such phosphate groups to proteins.
A 3D schematic representation of cyclin a protein. This protein partners with CDK2, for S and G2 stages of the cell cycle. Image credit: Wikimedia Commons.
Each stage of the cell cycle has a specific cyclin/CDK pair that behaves as a single unit, called a complex. Each stage's cyclin/CDK complex is required to allow the cell to commit to and proceed to the next stage of the cell cycle. When activated by a cyclin, a CDK is able to phosphorylate key proteins involved in the networks of chemical reactions in the cell, which in turn are activated and proceed to manufacture the cyclin required for proceeding into the next stage of the cell cycle. It is a beautifully elegant system of control, and it works to ensure that cells grow and divide when they're supposed to, and remain quiescent at all other times.
Antigrowth signals are proteins, exactly like the growth factors that I mentioned in the previous Hallmarks of Cancer article, except that they inhibit growth rather than promote growth. Antigrowth factors can therefore block cell growth by the two mechanisms mentioned above: cellular quiescence through G0 and the post-mitotic state. A cancer cell must therefore evade these signals if it is to continue dividing uncontrollably. During the G1/S Checkpoint, cells monitor their microenvironment to choose whether to continue in the cell cycle, enter G0 or enter a post-mitotic state. At the molecular level, nearly all antigrowth signals are funneled through a protein known as the Retinoblastoma protein. Therefore, Retinoblastoma is classified as a tumor suppressor protein. Indeed, it was the very first tumor suppressor protein to be discovered in 1971 through an elegant process of deduction and statistical analysis of rare eye cancers.
Retinoblastoma Protein
If a cell detects that it has damaged DNA at the G1/S checkpoint, this damaged DNA should not be replicated. Therefore, at the G1/S checkpoint, prior to entry into S stage, the brakes are slammed and everything comes to a screeching halt. In this analogy, the Retinoblastoma protein acts as the brake. Retinoblastoma protein is active when it is not phosphorylated, meaning phosphorylation inactivates Retinoblastoma. A primary function of Retinoblastoma is to bind to and inactivate E2F transcription factors. These are extremely important proteins that bind to DNA and activate genes which...you guessed it, control the cell cycle and DNA replication, including the Cyclins and CDKs specific to G1 and S phases of the cell cycle. In other words, E2F transcription factors are controlled by their interaction with the Retinoblastoma protein, which acts as the brake in the cell cycle progression to the S Stage. During G1, Retinoblastoma protein binds to E2F and blocks the production of S stage Cyclins and CDKs. When cells are stimulated to divide by external signals in their microenvironment, active CDKs specific to the G1 stage accumulate and phosphorylate Retinoblastoma. The Retinoblastoma protein, now inactivated, moves away from E2F, allowing the cell cycle to proceed. The brake is disengaged (see diagram below).
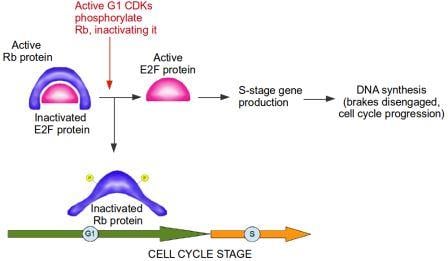
Active RB (retinoblastoma protein) inactivates E2F protein. CDKS (cyclin-dependent kinases) from G1 stage of the cell cycle phosphorylate RB, thereby inactivating it. E2F is now active and free to produce genes required for progression of cell cycle into s stage. The brake, from RB, is disengaged. Image credit: Buddhini Samarasinghe
Without E2F transcription factors, cell division grinds to a halt, and the phosphorylation status of Retinoblastoma controls the activity of these E2F transcription factors! Think about it for a moment: the addition or removal of a tiny molecule of phosphate to a single Retinoblastoma protein within the cell is responsible for whether a cell divides or not!
What influences the activation (i.e. phosphorylation status) of the Retinoblastoma protein? Antigrowth signals from the surrounding microenvironment of the cell. Given how important a role Retinoblastoma plays in the control of cell division, it comes as no surprise that cancer cells have found a myriad ways to bypass these antigrowth signals. Disruption of the retinoblastoma pathway liberates E2F transcription factors to promote cell division, and thus cells become insensitive to antigrowth signals that normally control this process.
Antigrowth signals
So what are these antigrowth signals? Probably the best documented is the signaling molecule TGF-beta. Recall from the previous paragraph that phosphorylated Retinoblastoma is inactive, and this 'disengages' the brakes on the cell cycle. TGF-beta has many different mechanisms for preventing the phosphorylation of Retinoblastoma (i.e. for preventing the disengagement of the brakes). Therefore, the presence of TGF-beta blocks the advancement of the cell cycle. Unsurprisingly, many cancers target this pathway for disruption. Some cancer cells stop responding to TGF-beta altogether, by producing less TGF-beta receptors on their cell surfaces. Other cancers produce mutated receptors that do not respond to the presence of TGF-beta. Some cancers get rid of downstream proteins that respond to TGF-beta. In many late-stage tumors, instead of functioning as an antigrowth signal, TGF-beta activates a cellular program known as Epithelial-to-Mesenchymal-Transition (EMT), which gives cancer cells stem-cell like abilities that is really bad news to a cancer patient. Finally, Retinoblastoma protein itself, the end target of this pathway, can be lost through mutation of its gene. Interestingly, certain cancer-promoting proteins (oncoproteins) can block the function of Retinoblastoma as well. For example, the human papillomavirus produces a protein known as E7, which binds to and inactivates Retinoblastoma.
The end result is that antigrowth signals, funneled through Retinoblastoma protein into the cell cycle, are, in one way or the other, disrupted in a majority of human cancers. Cancer cells with defects in the Retinoblastoma pathway are missing the services of a critical 'gatekeeper' of cell cycle progression; the absence of the Retinoblastoma gatekeeper permits persistent cell division. Therefore, the insensitivity to antigrowth signals represents a vital breach of an anti-cancer defense mechanism that is hardwired into our cells.
Reprinted with permission of Buddhini Samarasinghe and Scientific American, September 25, 2013
3. Evading Apoptosis
Apoptosis is the opposite of cell growth; it is cell death. To divide and grow uncontrollably, a cancer cell not only has to hijack normal cellular growth pathways, but also evade cellular death pathways. Indeed, this acquired resistance to apoptosis is characteristic of all types of cancer. But before I explain how cancer cells do this, we need to understand how the process of cellular death occurs in a normal cell.
The apoptotic program is hardwired into every single cell in our body. It is like a cyanide capsule, swiftly delivering death if the circumstances require cellular suicide. If a cell detects that it has damaged DNA, it can activate apoptosis to remove itself from the population. Apoptosis, or cellular suicide, is an entirely normal function of cells. The same apoptotic program is activated when a tadpole changes into a frog; the cells in the tail die through apoptosis, and the tail disappears. The same is true for the webbing between our fingers in our early embryonic development. Apoptosis is an extremely tidy process; cellular membranes are disrupted, the chromosomes are degraded, the DNA breaks up into fragments, and the dying, shrinking cell is swallowed up by a neighboring cell or a patrolling immune cell, leaving no trace of the cellular suicide behind.
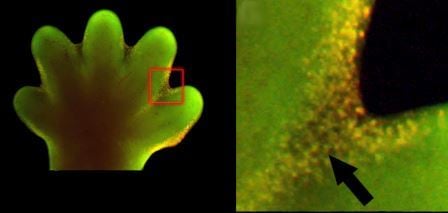
Apoptosis is also observed in normal embryonic development, not just for cancer. On the left is the paw of a mouse embryo, 15 days after fertilization. On the right is an enlarged view of the same paw, showing enhanced apoptosis in cells between the webbing of the fingers (the glowing green dots are apoptotic cells). Image credit: Amitabha Bandyopadhyay et. al., 2006 Plos Genetics DOI:10.1371/journal.pgen.0020216 (http://goo.gl/vkdaqa)
Regulators and Effectors
So how does apoptosis work at the molecular level? The apoptotic machinery can be divided into two broad categories: regulators and effectors. The regulators are responsible for monitoring the interior and exterior environment of the cell for conditions of abnormality in order to decide whether that cell should live or die. The possible abnormalities include DNA damage, signaling imbalance caused by the activation of cancer causing genes (oncogenes), lack of an oxygen supply or insufficient growth factors.
Apoptosis can therefore occur either through an intrinsic pathway, in which signals from within the cell activate the process, or through an extrinsic pathway where death signals from outside the cell are received and processed by the cell to activate apoptosis. It is thought that the intrinsic apoptotic pathway is more important in cancer prevention than the extrinsic pathway. Given how our cells carry machinery to destroy themselves with the precision of an executioner, it comes as no surprise that the process is tightly regulated.
The primary regulators of apoptosis are proteins belonging to a group known as the Bcl-2 family. These proteins can either be pro-apoptotic or anti-apoptotic; Bcl-2, Bcl-XL, Bcl-W, Mcl-1 and A1 proteins function as anti-apoptotic proteins that inhibit apoptosis, while Bax, Bad, Bid, Bok, Bik and Bak (I swear these names are not made up!*) are pro-apoptotic proteins that trigger apoptosis when activated. The anti-apoptotic proteins bind to and inactivate the pro-apoptotic proteins in a healthy cell that does not need to die. Apoptosis regulators also include death receptors on the cell surface which bind to death signaling molecules, as part of the extrinsic apoptotic pathway. This is similar to the way growth factors bind to and activate growth factor receptors, as I described previously, and this binding triggers the effectors of apoptosis.
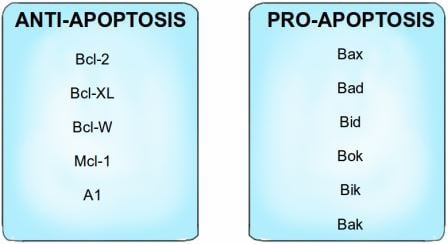
The Bcl-2 family of proteins can be divided into pro-apoptotic proteins and anti-apoptotic proteins, i.e. proteins that promote apoptosis and proteins that inhibit apoptosis. *For an explanation of the 'interesting' names for the pro-apoptotic proteins, see note at the end of this article! Image credit: Buddhini Samarasinghe
The Suicide Machines
Where is Mission Control for apoptosis? Many of the apoptotic signaling pathways converge at the mitochondria. Mitochondria are tiny organelles floating within a cell, and function as the cell's energy factories. They contain a signaling molecule known as cytochrome c, which is bound to the mitochondrial membrane. In response to pro-apoptotic signals (from pro-apoptotic proteins such as Bax), cytochrome c is released into the cell by the mitochondria, and they bind to a protein known as Apaf-1. This results in the formation of the apoptosome. The apoptosome is an extremely beautiful structure resembling a wheel with seven spokes. Once formed, the apoptosome goes on to activate a group of proteins known as caspases.
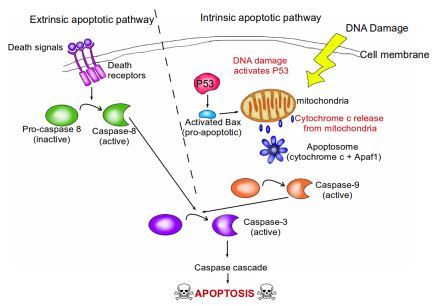
A brief overview of the extrinsic and intrinsic apoptotic pathways. In the extrinsic pathway, death signals from the surrounding environment of the cell bind to death receptors on the surface of the cell membrane. This causes the conversion of inactive pro-caspase-8 into active caspase-8. Caspase-8 then goes on to activate caspase-3, which begins the caspase cascade that leads to apoptosis. In the intrinsic pathway, typically initiated by DNA damage, P53 is activated. P53 then activates the pro-apoptotic protein bax, which initiates the release of cytochrome C from the mitochondria. APAF-1 and the released cytochrome C combine to form a complex known as the apoptosome. The apoptosome causes the conversion of inactive pro-caspase-9 into active caspase-9. Caspase-9 then goes on to activate caspase-3 in a similar manner to the extrinsic pathway. Activated caspase-3 then leads to the caspase cascade, resulting in apoptosis. Image credit: Buddhini Samarasinghe
All our cells contain the seeds of their own destruction; these come in the form of caspases. Caspases can be thought of as cellular executioners. They are proteins that degrade other proteins in our cells. Active caspases can wreak havoc within a cell and are therefore extremely dangerous, so they are produced in an inactive form by the cell (known as pro-caspases), like an executioner's blade that is sheathed. Upon detecting an increase in the amount of cytochrome c, released from the mitochondria, the blades are unsheathed. There are 13 such caspase genes identified in the human genome so far. Two of the caspase proteins act as 'gatekeeper' caspases: caspase-8 and caspase-9. They are initiator caspases that, when activated by cytochrome c release, go on to activate the other caspases in a cascade of irreversible cellular protein degradation.
P53: The Guardian of the Genome
How do cells detect the necessary conditions for triggering apoptosis? In the previous Hallmark of Cancer article, I explained the fundamental role of the Retinoblastoma protein in controlling cell division. Retinoblastoma, remember, is a vitally important brake on cell division. Damage to a cell's Retinoblastoma gene releases this brake, leading to uncontrolled cell growth.
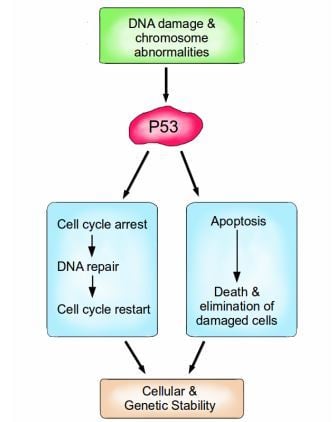
P53 is a vital protein; more than half of all cancers have inactive p53. When activated by either DNA damage or chromosome abnormalities, p53 can halt the cell cycle and initiate DNA repair. If repair is successful, the cell cycle is restarted, and cellular and genomic stability is re-established. If repair is not an option because the damage is too great, Then P53 can facilitate apoptosis, leading to the death and elimination of the abnormal cell. Image credit: Buddhini Samarasinghe
Similarly, P53 is an extremely important protein, dubbed 'The Guardian of the Genome'. Amongst its many functions, it is responsible for detecting DNA damage, chromosome abnormalities and arresting the cell cycle to initiate repair; if repair is not possible then apoptosis is induced. P53 induces apoptosis by increasing the production of the pro-apoptotic protein Bax. Bax stimulates the mitochondria to release cytochrome c, which activates the caspase cascade which ultimately results in cell suicide. P53 is vital for maintaining the integrity of our genome at the most fundamental level.
Evading apoptosis
So how do cancer cells escape death? The most common method is the loss of the apoptosis gatekeeper, the protein P53. More than half of all types of human cancers have a mutated or missing gene for p53, resulting in a damaged or missing P53 protein. As an alternative to achieving the loss of P53, cancer cells can compromise the activity of P53 by increasing the inhibitors of P53, or silencing the activators of P53. Previously I explained how the human papillomavirus produces a protein known as E7, which binds to and inactivates Retinoblastoma.
Similarly, another protein, E6, also produced by the human papilloma virus, binds to and inactivates P53. These two cancer-causing proteins, E6 and E7 (oncoproteins), therefore disable two vital gatekeepers, Retinoblastoma and P53, that control both cell division and cell death; the result is repeated uncontrolled cell division that manifests itself in warts, with strong associations with the development of cancer. Cancer cells can also produce excessive amounts of anti-apoptotic proteins such as Bcl-2, Bcl-XL etc. They can produce less of the pro-apoptotic proteins such as Bax and Bak. They can short-circuit the extrinsic death receptor apoptotic pathway.
It comes as no surprise that highly aggressive cancers often have both Retinoblastoma and P53 mutations. As a result, these rapidly growing tumors have extremely low levels of apoptosis and extremely high levels of cell division. Like de-rigging the Sword of Damocles, cancer cells can inactivate the machineries of death; and the evasion of apoptosis by cancer cells therefore represents a key breach of an extremely important anti-cancer defense mechanism.
*in case you were curious, the pro-apoptotic members of the Bcl-2 family are thus named:
Bax: Bcl-2 Associated X protein
Bad: Bcl-2 Associated Death promoter
Bid: BH3 interacting-domain Death agonist
Bok: Bcl-2 related Ovarian Killer
Bik: Bcl-2 Interacting Killer
Bak: Bcl-2 homologous Antagonist Killer
Reprinted with permission of Buddhini Samarasinghe and Scientific American, October 2, 2013
4. Limitless Replicative Potential
The Fourth Hallmark of Cancer is defined as "Limitless Replicative Potential".
The first three Hallmarks of Cancer explain how independence from growth signals, insensitivity to antigrowth signals and resistance to apoptosis lead to the uncoupling of a cell's growth program from the signals in its environment. However, cancer is not just a result of disrupted signaling. Our cells carry an in-built, autonomous program that limits their multiplication, even in the face of disrupted signals from their environment. For a single cancer cell to develop into a visible tumor, this program must also be disrupted.
The Cellular Timekeeper
Normal cells are hard wired with a timer that keeps track of their age; the number of times they divide and grow. Most cells in our body can only undergo a limited number of successive cell growth-and-division cycles. This limit is named the Hayflick Limit after its discoverer, Leonard Hayflick. After undergoing between 40 and 60 divisions, cell growth slows down and eventually stops altogether. This state is known as senescence, and it is irreversible; although the cell does not grow or divide, it remains alive. When normal human cells are cultured in the lab in a petri dish, we can observe this phenomenon, where cells grow and divide a fixed number of times and then enter senescence. Some cells are able to make it past the senescence barrier and continue dividing; however these cells then undergo a second phenomenon known as crisis, during which the ends of their chromosomes fuse with each other, and the cells all die on a massive scale via apoptosis.
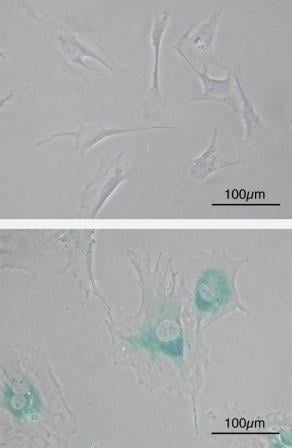
Microscope image of normal cells (upper) compared to senescent cells (lower). Blue-green areas indicate expression of marker associated with senescence. Image credit: Wikimedia Commons.
How does a cell count its divisions? How does it 'know' when to stop? The answer is telomeres. Telomeres are regions of repetitive DNA, capping and protecting the ends of the chromosome from degrading or from fusing with another chromosome. Without telomeres, each time a cell divides our genomes would progressively lose information because the chromosomes would get shorter and shorter. A telomere is like the heat-shield of a spacecraft; it protects the actual spacecraft and absorbs the damage instead. With every replication of a cell, about 50-100 nucleotides of telomeric DNA is lost. This progressive loss eventually causes the telomeres to lose their ability to protect the ends of chromosomal DNA. Left unprotected, these exposed ends become damaged. The DNA damage response is activated, leading to growth arrest; senescence. When chromosome ends fuse with each other, this irreversible damage results in the activation of apoptosis; the cell enters crisis, and dies.
The End Replication Problem
Why do the ends of chromosomes shorten? To understand this, first we need to go over the basic mechanisms of DNA replication. A cell must replicate its DNA before it divides. DNA is a double-stranded molecule, and each strand of the original DNA molecule serves as a template for the production of a complementary strand. The two strands have a directionality; the two ends of a single strand are known as the 3' end and the 5' end. The numbers refer to the position of the carbon atom in the deoxyribose molecule at the end of the strand to which the next phosphate molecule in the DNA chain attaches. For a quick introduction into the structure of DNA, check out this YouTube video:
This directionality matters because DNA replication takes place under the direction of an enzyme known as DNA polymerase. This enzyme faithfully copies our genetic code letter by letter. However, DNA polymerase can only work in one direction; the 5' to 3' direction.
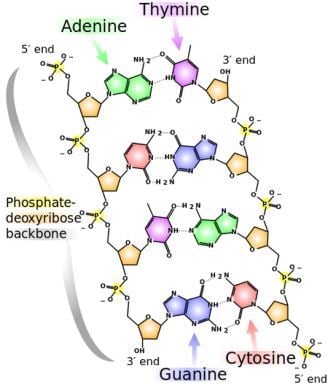
The chemical structure of DNA. The 5' and 3' ends are shown lying antiparallel to each other. Nitrogenous bases adenine (green), thymine (purple), guanine (blue) and cytosine (red). These bases are fixed to the phosphate-deoxyribose backbone. Image credit: Wikimedia Commons.
Therefore, although DNA replication is straightforward for one of the DNA strands (the 5' to 3' strand), the other strand (the 3' to 5' strand) is more complicated. This strand is replicated in short fragments instead of one continuous strand of DNA. Replication begins with an enzyme known as primase, which reads the template DNA and initiates the synthesis of very short complementary RNA fragments. DNA polymerase is now able to use these RNA fragments as a starting point to synthesize complementary fragments of DNA in between the RNA fragments. The RNA fragments are then removed and replaced with DNA, and the fragments of DNA are joined together by another enzyme, DNA ligase. This solves the directionality problem of DNA polymerase, but now we run into another problem, known as the end replication problem. This is because although the 5' to 3' strand can be replicated to the very end, the 3' to 5' strand cannot; DNA polymerase enzyme requires RNA fragments to begin replication, and there is nothing for such a fragment to attach to at the very end of the DNA strand. Therefore, with every round of replication, a small fragment of the DNA would be lost from the end of the chromosome, since it cannot be replicated. The cell solves this problem by having telomeres at the ends of chromosomes, where they prevent the loss of valuable genetic information by acting as a disposable buffer. Over time, with each successive round of DNA replication, the telomeric DNA shortens until finally there is no more disposable buffer, at which point the cell stops dividing and enters senescence. For a visualization of the end replication problem check out this YouTube video:
When cells are grown in petri dishes in the lab, repeated cycles of cell division lead first to senescence and then, for those cells that make it past this barrier, to crisis phase. Fascinatingly, in very rare instances (about 1 in 10X7) a cell can emerge from this ordeal exhibiting unlimited replicative potential. This cell is now said to be immortalized, and it is a trait that most cancer cells growing in labs exhibit, including the famous HeLa cells.
Telomere Maintenance
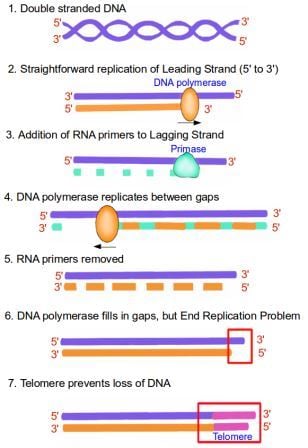
1. Double stranded DNA is made up of two DNA strands lying antiparallel to each other, with their 5' and 3' ends highlighted in red. 2. When DNA replicates, the two strands unwind. Replication of the leading strand is very straightforward, with the DNA polymerase enzyme copying from the purple template strand to form the new DNA strand, shown in orange. This new strand is formed in the 5' to 3' direction, since DNA polymerase only works in that direction. 3. For the synthesis of the lagging strand, the enzyme RNA primase creates short complementary RNA fragments, shown in green. 4. DNA polymerase is now able to use these short RNA fragments as a starting point to synthesize DNA between the RNA gaps, shown in orange. The enzyme still works from 5' to 3', in a 'backstitching' manner. 5. The RNA primers are now removed, and the gaps are filled in by DNA polymerase to form a complete replicate of the lagging strand. 6. However, the extreme end of the DNA strand cannot be replicated because there is nowhere for the DNA polymerase enzyme to attach to. 7. The presence of a telomere, shown in pink, allows the DNA to be protected as the telomere gets shortened instead. Image credit: Buddhini Samarasinghe
Cancer cells have therefore not only uncoupled their growth program from the signals in their environment, they have also breached the in-built replication limit hard wired into the cell. How do they achieve this? All cancer cells maintain their telomeres. 90% of them do so by increasing the production of an enzyme known as telomerase. As its name implies, telomerase functions by adding telomeric DNA to the ends of chromosomes. Most normal cells do not divide frequently, and therefore are not in any danger of shortened telomeres; these cells can get away with having low telomerase activity. Indeed, most cells apart from fetal cells and stem cells show low telomerase activity levels. Many cancer causing proteins (oncoproteins) are able to activate the production of telomerase, while many cancer preventing proteins (tumor suppressors) such as P53 (see previous Hallmark) produce factors that inhibit the production of telomerase. The other 10% of cancers rely upon the activation of a pathway known as the Alternative Lengthening of Telomeres (ALT), which swaps around telomeres to lengthen them.
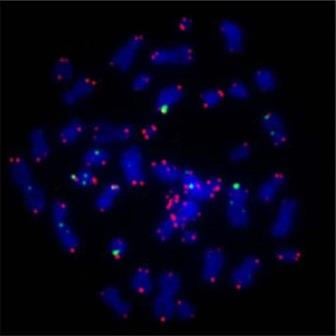
Human chromosomes, stained blue, with marker for telomere region in red. Image credit: Asako J. Nakamura (wikimedia.com)
Intriguingly, telomere length is also affected by oxidative stress. Oxidative stress, in the form of free radicals, damages DNA. This damage is usually repaired by DNA repair mechanisms, but these mechanisms are less effective on telomeric DNA than elsewhere on the chromosome. Telomeres are therefore highly susceptible to oxidative stress. It also explains the rate of telomere shortening observed; estimated loss per cell division because of the end replication problem has been estimated at 20 base pairs of DNA, yet the observed loss is much larger, between 50-100 base pairs of DNA. This difference shows that oxidative stress has a far greater impact on telomere length than the nuances of DNA replication. It is possible that cell senescence induced by telomere loss is therefore a stress response, evolved to block the growth and replication of cells that have been exposed to a high risk of DNA damage. The defining feature of a cancer cell is its ability to divide endlessly, without exhaustion, generation after generation. They achieve this by destroying the cellular timekeeper, the telomere. Immortality comes at a price; the accumulation of damaging mutations only increases with time, which is why cancer is primarily a disease of an aging population. The immortalization of cancer cells by telomere maintenance therefore represents an essential step in tumor progression.
Reprinted with permission of Buddhini Samarasinghe and Scientific American, October 9, 2013
5. Sustained Angiogenesis
In a developing embryo or a healing wound, communities of cells organize themselves into tissues, undertaking specialized tasks beyond the ability of any single cell to accomplish. These tissues require oxygen and nutrients, as well as a facility to remove metabolic wastes and carbon dioxide. The formation of new blood vessels, known as angiogenesis, satisfies these needs. In a similar manner, a growing tumor, an aggregation of cancer cells, also requires access to oxygen, nutrients and waste disposal. Beyond a certain size, typically 1 mm, diffusion alone is insufficient for providing these necessities; the surface area to volume ratio becomes too low and the developing tumor begins to starve. In response, cancer cells send out signals to the cells of nearby blood vessels, inducing these innocent bystanders to grow extensions to form a supply chain and drainage channels. Thus, cancer cells subvert these normal neighboring cells into playing a key role in driving tumor development.
The Angiogenesis Switch
Angiogenesis is a normal, essential process in the formation and development of embryonic tissues. In the course of normal development, the arrangement of our blood vessels is fixed; angiogenesis is switched off. In adults, angiogenesis is only switched on during physiological processes such as wound healing or menstruation, and then only transiently and regulated extremely carefully. In contrast, cancer cells always have angiogenesis switched on.
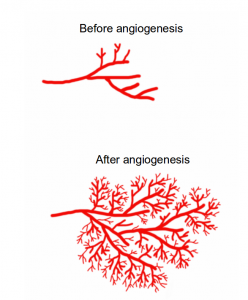
Angiogenesis is the formation of new blood vessels. In this image, angiogenesis causes blood vessels to sprout more branches. Image credit: Buddhini Samarasinghe
Regulation of Angiogenesis through VEGF
How is angiogenesis regulated? Counterbalancing positive and negative signals circulate in the cellular microenvironment surrounding every cell, respectively encouraging or blocking angiogenesis. These signals include growth factors, which I described previously, the primary angiogenic growth factor being Vascular Endothelial Growth Factor (VEGF). VEGF binds to VEGF receptors found on the surface of endothelial cells, which are cells that line the interior of blood vessels. VEGF causes endothelial cells to first break through existing blood vessels, then migrate towards the signal (i.e. VEGF), grow and replicate and form new blood vessels as directed. Negative angiogenic signals include the prototypical thrombospondin-1 protein, which inhibits the growth and migration of endothelial cells. The binding of thrombospondin-1 to endothelial cells triggers the activation of caspases, leading to apoptosis of the cell, as described previously. Currently we know of dozens of proteins that induce or inhibit angiogensis through similar mechanisms.
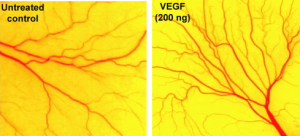
Angiogenesis assay using a chick embryo membrane. There are more blood vessels in the sample treated with VEGF (right) compared to the untreated control (left). Image credit: Chew-Voon Chin (Cancer Letters, Volume 283, Issue 1, 2009).
Hypoxia and Angiogenesis
How do cancer cells activate the angiogenic switch? Simply put, they change the balance of angiogenesis inducers and counteracting inhibitors in the surrounding cellular environment. The rapid growth of a tumor results in an oxygen delivery problem; cells found towards the inside of the tumor are deprived of an adequate supply of oxygen. These cells are now said to be hypoxic, and activate the hypoxia stress response. The primary response to hypoxia is mediated at the cellular level by the hypoxia inducible factor (HIF). HIF is a transcription factor, initiating the production of other proteins required to mediate the effects of hypoxia. Under typical oxygen levels, a hydroxyl (OH) molecule is added to HIF as a tag for degradation, and HIF levels are kept constantly low as a result. However, during hypoxia, this degradation halts and HIF accumulates within the cell. The accumulated HIF transports into the nucleus of the cell where it induces the expression of numerous target genes, including (you guessed it) VEGF. This increase in VEGF results in shifting the balance of angiogenesis inducers and inhibitors, thereby switching on angiogenesis.
By activating the angiogenic switch, tumors co-opt endothelial cells. In addition, tumors also recruit cells known as pericytes. These are support cells, providing structural support for the newly formed blood vessels, promoting the survival of the endothelial cells, and guiding the sprouting of new blood vessels. How do endothelial cells recruit pericytes? Again, it is through a cell signaling mechanism. Activated endothelial cells secrete a growth factor known as Platelet Derived Growth Factor (PDGF). Pericyte cell surfaces have PDGF receptors, which are activated upon the binding of PDGF. This makes the pericytes secrete more VEGF, resulting in a positive feedback loop, initiated by the tumor cell, resulting in the rapid growth of new blood vessels.
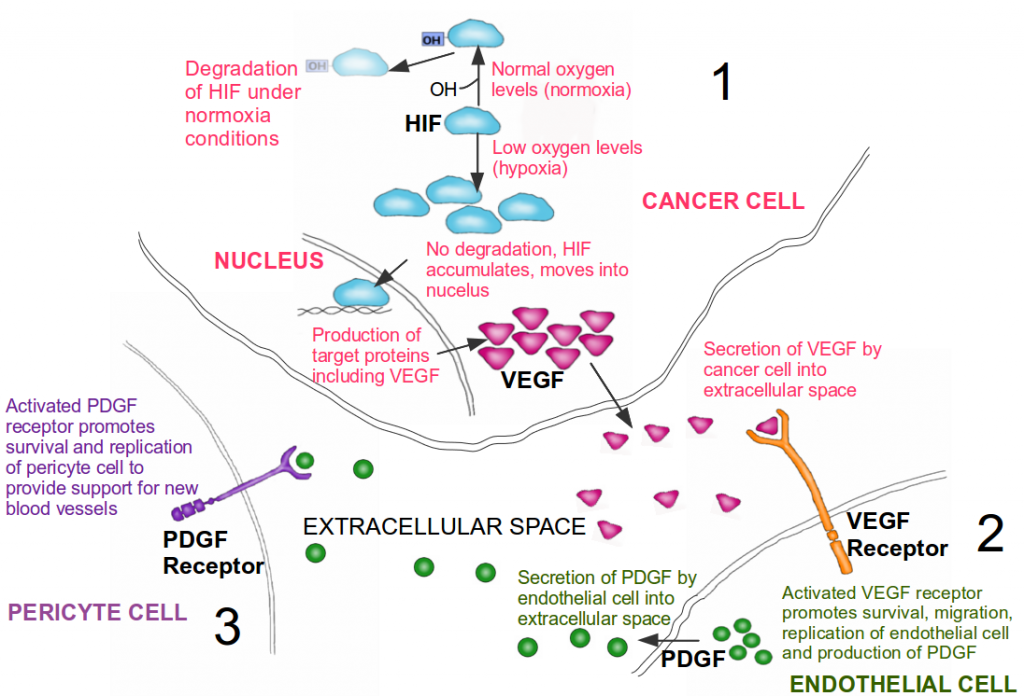
Activation of angiogenesis. 1. In a cancer cell, under normal oxygen conditions (normoxia) the HIF protein has a hydroxyl (OH) group added to it; this tags it for degradation. Under low oxygen levels (hypoxia), HIF is not degraded and it accumulates within the cell. HIF then moves into the nucleus where it activates the production of proteins involved in the hypoxia stress response pathway, including the growth factor VEFG (pink triangles). 2. VEGF now moves from the cancer cell into the extracellular environment where it binds to VEFG receptors on the surface of endothelial cells. This binding results in the survival, migration and replication of endothelial cells, ultimately promoting the formation of new blood vessels (i.e. angiogenesis). Endothelial cells also produce PDGF (green circles). 3. PDGF is secreted by endothelial cells into the extracellular space, where it binds to PDGF receptors found on the surface of pericyte cells. This binding results in the survival and replication of pericyte cells, which in turn lend structural support for the newly formed blood vessels. A cancer cell can thus subvert innocent bystander cells for its own needs. Image credit: Buddhini Samarasinghe
The key to angiogenesis is VEGF, and it comes as no surprise therefore that VEGF production is tightly regulated. The activation of cancer-causing proteins (oncoproteins) such as Ras results in an increase in VEGF production. Cancer cells also stop producing angiogenesis inhibitors. Thrombospondin-1, a potent angiogenesis inhibitor, is under the positive control of P53, a key protein involved in protecting our cells from cancer. Consequently, the loss of P53, found in more than half of all cancers, causes thrombospondin-1 levels to fall. It is an elegant system of subversion in which cancer cells hijack our normally tightly regulated pathways to serve their own ends.
Angiogenesis Inhibition
How important is angiogenesis for cancer progression? Many experiments have shown that proteins which block the action of VEGF were able to impair the growth of tumors in mice. Conversely, the addition of pro-angiogenic factors such as VEGF encourage tumor growth. This raises the obvious question of whether we could use these anti-angiogenesis factors (which occur naturally in our bodies) to fight back. Indeed, many such anti-angiogenesis factors are currently undergoing clinical trials in cancer patients. Avastin was the first commercially available angiogenesis inhibitor; it binds directly to VEGF and prevents it from binding to the VEGF receptor. Also of interest are endostatin, angiostatin and the previously mentioned thrombospondin-1. Small molecule drugs that can inhibit VEGF are in clinical trials; notably sorafenib, axitinib and pazopanib.
In normal tissues, communities of cells instruct each other to grow through signaling pathways. The tumor microenvironment hijacks these selfsame pathways to divide and grow uncontrollably. A cancer cell cannot function in isolation. It is dependent on its surrounding cells, the innocent bystanders that faithfully respond to the malevolent signals that cancer cells use to achieve their uncontrolled growth. The angiogenesis within the context of a tumor is a warped, twisted version of what it should be; the vessels are convoluted and excessively branched. They are distorted and enlarged, with erratic blood flow and leakage. Tumors are messy and bloody, a result of the angiogenesis that sustains them. Angiogenesis in a cancer is a perversion of a normal cellular process, a perversion that is an essential requirement for the development of cancer.
Reprinted with permission of Buddhini Samarasinghe and Scientific American, October 18, 2013
6. Tissue Invasion and Metastisis
A growing tumor will eventually spawn pioneer cells; these move out of the original clump of mutant cells to invade adjacent tissues and then travel to distant sites where they form new colonies. These distant settlements of cancer cells are named metastases and, with the exception of leukemias and some brain tumors, cause the majority of cancer deaths. Metastasis is bad news, with significantly reduced survival rates and prognosis for patients. The ability to metastasize allows cancer cells to find new areas of the body where space and nutrients are not limiting. How do cancer cells do this?
The Extra Cellular Matrix
In biology, a tissue is an aggregation of cells that performs a specific function. Tissues combine to form organs; organs combine to form a body. Our tissues are composed primarily of two types of cells; epithelial and mesenchymal cells. Epithelial cells adhere to one another to form cell layers, which act as barriers to protect our bodies and organs from the environment. In contrast, mesenchymal cells are solitary and capable of migrating. Our tissues are not made up solely of cells. A large proportion of tissue consists of extracellular space, which is filled with a mixture of carbohydrate and protein molecules; this space is known as the Extracellular Matrix (ECM). The molecules that make up the ECM are secreted by cells embedded in it, and these cells tether themselves to the ECM (and to one another) to form tissues. Metastasis therefore requires the untethering of these bonds, to allow predacious cancer cells to migrate freely.
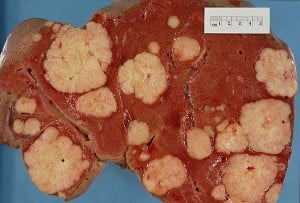
Cross section of a human liver, taken at autopsy examination, showing multiple large pale tumor deposits. The tumor is an adenocarcinoma derived from a primary lesion in the body of the pancreas. Credit: Haymanj/Wikimedia Commons
Several classes of proteins are involved in the tethering of cells to their surroundings. Immunoglobulins and cadherins mediate cell-to-cell adhesions while integrins link cells to the ECM. All of these interactions convey regulatory signals to the cell and should not be viewed as static connections that simply hold cells in place. The most important protein cementing cells to each other is known as E-cadherin. The coupling of cells by E-cadherin results in the transmission of antigrowth signals; this is one of the mechanisms for the phenomenon known as contact inhibition, where cells that touch one another do not grow any further. Unsurprisingly, E-cadherin function is lost in migrating cancer cells. Conversely, another molecule known as N-cadherin shows increased levels in migrating cancer cells, as this molecule helps the cancer cell to slip through blood vessels during migration.
What are the traits of migrating cancer cells? These cells change their appearance, from a neat, ordered cobblestone-like shape to spindly and long-limbed. The cells also untether themselves from the ECM, by expressing proteins that degrade the ECM, notably matrix metalloproteases. They stop expressing E-cadherin, so that the cement that binds them to other cells is eliminated. They express more N-cadherin, so they can move through blood vessels to distant sites more efficiently. A metastatic cancer cell has increased motility and is resistant to apoptosis. These traits occur through the activation of a cellular program that promotes migration, and the molecular mechanism for this program has been identified only recently, within the last decade.
Hijacking an Embryonic Program
What is the molecular mechanism for cell migration? Under normal circumstances, cells migrate during embryonic development and in response to inflammation. The primary pathway involved in this process is an important developmental program known as the epithelial to mesenchymal transition, or EMT. EMT is a cellular program that has been known in the context of embryonic development for many years. Epithelial cells can be defined as surface cells that display a distinct polarity, that is, knowing which way is up because of being tethered to one another and the underlying ECM. In contrast, mesenchymal cells are loosely packed, have no polarity and are able to migrate freely. During embryonic development, epithelial cells are able to undergo physical and genetic changes collectively referred to as EMT. This occurs through the degradation of the ECM, dissolution of E-cadherin and other cell adhesion junctions and the loss of cell polarity, resulting in the formation of migratory mesenchymal cells that have invasive properties. These cells are then recruited to specific sites in the developing embryo where they can revert back through MET (the reverse of EMT; mesenchymal to epithelial transition) to form epithelial tissues at distant locations.
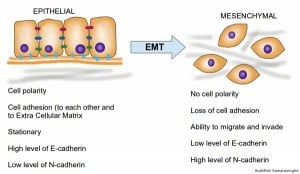
Epithelial to mesenchymal transition, with associated characteristics of the two cell types. Credit: Buddhini Samarasinghe.
Cancer cells hijack this process, first to enable metastasis through activating the EMT program and then, once they reach their new home, to revert back to epithelial form by activating the MET program. Thus although the EMT-MET program has been known of in the context of embryonic development for many years, it is only recently that the significance of this EMT-MET program to cancer progression has been recognized.
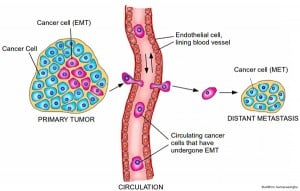
EMT and metastasis. A tumor will have a population of cells that have undergone EMT. These cells are now able to migrate freely, by entering the circulatory system. Upon reaching their final destination, the cells undergo the reverse MET program and forms a secondary tumor at a distant site. Credit: Buddhini Samarasinghe.
Intriguingly, tumors can also possess cancer stem cells; these cells are highly capable of forming tumors. They are thought to persist in tumors as a distinct population of cells and cause relapse and metastasis by forming new tumors. Even a single cancer stem cell can give rise to an entire tumor and worryingly seem resistant to chemotherapy. Recent evidence indicates that a cancer cell that has undergone EMT is, in fact, a cancer stem cell.
TGF-beta: The Ying and the Yang
How do cancer cells activate EMT? In a previous article I explained how the signaling molecule TGF-beta acts as an antigrowth factor, by blocking the cell cycle. But TGF-beta has a darker side; while it acts as a tumor suppressor in the early stages of cancer, it can contribute to malignancy by promoting invasion and metastasis by activating the EMT program. Indeed, the addition of TGF-beta to epithelial cells in culture is a very handy method for inducing the EMT program. The EMT program has a dedicated set of transcription factors; proteins that activate the production of yet more proteins, in a now familiar signaling cascade, to enable the shift from epithelial to mesenchymal form. These transcription factors are Snail, Slug, Twist and ZEB1/2. The signaling cascade is as follows; binding of TGF-beta to its receptor phosphorylates two proteins known as Smad2 and Smad3. These two activated proteins then combine with Smad4, to form a kind of protein 'threesome' that moves into the nucleus, where it activates the production of Snail/Slug, ZEB1/2 and Twist. These transcription factors repress the expression of epithelial characteristics, while activating the expression of mesenchymal characteristics. TGF-beta can also function through non-Smad pathways, all towards shifting the cell's characteristics from epithelial to mesenchymal form. Most important, the activation of these proteins also result in the repression of E-cadherin production; the cement mixer grinds to a halt.
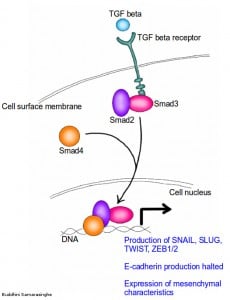
TGF beta signaling pathway for the induction of EMT. TGF beta binds to TGF beta receptor on cell surface. This activates Smad2 and Smad3 proteins, which go on to combine with Smad4. The resulting Smad group of proteins then moves into the cell nucleus and begins the production of snail, slug, twist and ZEB1/2 proteins. These proteins now halt the production of E-cadherin and enable the expression of mesenchymal characteristics. Credit: Buddhini Samarasinghe.
It might seem difficult to reconcile how TGF-beta can play both good cop and bad cop in cancer progression. The explanation is, again, beautiful in its simplicity; cancer cells often have mutations in the TGF-beta signaling pathways that inhibit growth, thus making room for those other pathways where TGF-beta aids in cancer progression.
More Innocent Bystanders
Metastasis and invasion are complicated processes, and these topics continue to be very active fields of research. In the previous articles, I explained that cancer cells further their uncontrolled growth by ruthlessly co-opting their normal neighbor cells to breach the anti-cancer defense mechanisms represented by these hallmarks. This is also true of metastasis. Macrophages, a type of immune cell, turn traitor and contribute to tumor growth and progression. These cells are attracted to the edges of the tumor and supply it with enzymes to degrade the ECM, to enable the cancer cells to break free and migrate. These tumor-associated macrophages also supply growth factors to cancer cells, to enable them to divide and grow; cancer cells reciprocally stimulate the macrophages by producing their favorite growth factor, known as Colony Stimulating Factor. Thus cancer cells can secrete various signaling molecules that recruit pro-invasive inflammatory cells to do their dirty work for them, rather than producing enzymes to degrade the ECM themselves.
Metastasis has been traditionally thought of as the final stages of a burgeoning tumor; indeed, our current classification system for a tumor upon diagnosis assigns a higher value when there is evidence of metastasis. We used to think that cancer cells pass through multiple successive mutations, growing in size, eventually sending away migratory cells to set up shop elsewhere. Dismayingly, recent evidence suggests that metastasis does not necessarily happen in the final stages of cancer progression, but can occur at any time--sometimes even before the primary tumor is observed. The ability to invade and metastasize distant sites is a key hallmark of cancer progression and possibly one of the most complicated and least understood hallmarks so far.
Reprinted with permission of Buddhini Samarasinghe and Scientific American, October 30, 2013
7. Genome Instability and Mutation
Not all cancer cells are equal. They vary, they compete, and the fittest survive to pass on their genes to daughter cells, which continue to vary, compete and survive. If this sounds familiar, it's because it is; cancer cells evolve. Within the complex ecosystem that is our body, cancer cells mutate and face selective pressures as they change and adapt to their environment. The evolving cancer cell has to out-compete the normal cells surrounding it, evade attack by immune cells, escape the apoptotic machinery which causes cells to self-destruct, corrupt and co-opt otherwise loyal surrounding cells and migrate to distant parts of the body.
Sometimes, when the sequences of nucleotides called genes mutate, those mutations are not corrected. It doesn't happen often (each nucleotide has only a one-in-a-hundred-billion to a one-in-a-billion chance of being miscopied), but each time a cell divides and passes a copy of its DNA to its daughter cell, the chance of error increases. An average gene comprises about 1,700 nucleotides. Over one hundred million billion cell divisions occur over the average human lifetime. The average gene is thus home to somewhere between one hundred million and ten billion mutations, spread over its copies in each cell. If just one of those mutations increases the evolutionary fitness of its cell, then that mutation will expand into many cells, increasing the probability that subsequent mutations will build on the first.

Mutations are inevitable. While mutations are still rare events, the sheer number of opportunities (i.e. cell divisions) for mutations during an average human lifetime makes the probability of a mutation for an average gene high. Image credit: Buddhini Samarasinghe
We know that cancer occurs because we can see the stepwise accumulation of mutations. If a cancer cell acquires a mutation that enables it to grow faster, or survive longer and produce more offspring than the surrounding cells without that mutation, then that cancer cell has a selective advantage. Its descendants will be fitter; they will outgrow and dominate the local tissue environment. These principles of population genetics and evolutionary biology describe the process of tumor formation and are key to understanding how cancer cells can become resistant to drug treatment; they evolve that resistance.
Mutations 101
Mutations enable cancer cells to embark on their frenzied growth within our bodies. But what are mutations, and how do they happen? In genetics, a mutation is a change in an organism's DNA sequence. The nucleotide letters A, T, C and G that make up our DNA can be deleted or substituted, and single or double stranded breaks can occur in the DNA molecule. Complete sections of our DNA can also be deleted or swapped with other sections. These changes can occur spontaneously or from exposure to inducers such as harmful chemicals or radiation. Our metabolic activities cause mutations all the time; oxygen, the vital molecule that helps us live, also creates dangerous DNA-damaging free radicals when metabolized by our cells. A sunny day at the beach can introduce thousands of mutations into our DNA. In fact, mutations are inevitable; each time our cells divide, the imperfect DNA replication process introduces temporary errors into our DNA. It has been estimated that all these processes can result in thousands of individual molecular lesions per cell per day. Our genome surveillance system and DNA repair mechanisms must be doing a fantastic job: based on these mutation rates, cancer should occur from the moment we are conceived.
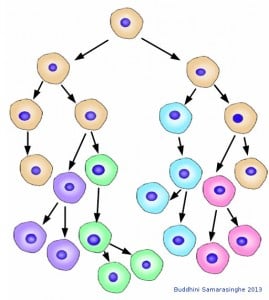
Cancer cells evolve. With each cell division, cancer cells accumulate mutations that cause variations. These variations allow cancer cells to change and adapt to survive within the complex ecosystem that is our body. In this schematic, the original cancer cell type shown in beige is no more; its descendants have all mutated and diverged, adapting and surviving with each cell division. Image credit: Buddhini Samarasinghe
What is our genome surveillance system? If a cell is to survive, all the different mutations must be repaired. Typically, this repair involves cutting out and re-synthesizing the damaged portion of the DNA. Although there are many different types of DNA repair, all are so important that the proteins involved can be found in everything from the humblest bacterium to our own cells.
DNA Repair Mechanisms
The beauty of DNA repair comes from the structure of DNA: The double helix carries two separate copies of all the genetic information in each of its two strands. When one strand is damaged, the other is used as a template to restore the sequence to the damaged strand. When both strands of the double helix are broken, however, leaving no template strand for repairs, cells may use one of two distinct alternative mechanisms to address breaks. The first, known as Non-Homologous End Joining (NHEJ), is a quick and dirty method for fixing the break; simply put, the two broken ends are brought together and joined, usually with the loss of one or two nucleotide letters at the site of joining. Although this loss results in a permanent change in the DNA sequence, it is less harmful for the cell than a persisting double-stranded break. And, since very little of our genome actually codes for protein, the resulting mutation is statistically unlikely to be of much consequence. In contrast to the error-prone NHEJ mechanism, Homologous Recombination DNA Repair exploits the fact that each cell contains two copies of each double helix (i.e. each cell has two copies of each chromosome). In this case, the repair mechanism is able to transfer nucleotide sequence information from the intact double helix to the broken one. Recombination proteins recognize regions of similarity between the two double helices and bring these regions together. Then, DNA replication proteins use the intact helix as a template to repair the broken one.
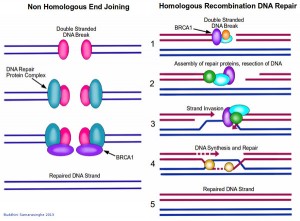
Double stranded DNA breaks are repaired by two main mechanisms: non homologous end joining (NHEJ, left), and homologous recombination DNA repair (right). BRCA1 (purple) plays an important role in both these mechanisms. In NHEJ, a double stranded DNA break is detected by DNA repair proteins which bring together the two ends and join them. This method usually results in the loss of several nucleotides, and is error prone as a result. Homologous recombination DNA repair is more stringent. Since each cell has two copies of each chromosome, this mechanism uses one copy as a template to repair a double stranded break in the other. 1. Repair proteins assemble at the site of DNA damage. The damaged strands are indicated in red while the intact DNA strands are indicated in blue. 2. The repair proteins resect the DNA to create some room to work in. 3. The intact copy of DNA (blue strands) then 'invades' the damaged strand. 4. Repair proteins are able to use this intact copy of DNA as a template to repair the break in the damaged strand, by synthesizing DNA across the gap. 5. The double stranded DNA break is repaired, with a high level of fidelity. Image credit: Buddhini Samarasinghe
Molecular Caretakers: BRCA1 and BRCA2
Two of the most famous proteins in cancer, BRCA1 and BRCA2, play a central role in DNA repair. These proteins move to the site of DNA damage and recruit other proteins to initiate DNA repair complexes. When either BRCA1 or BRCA2 are absent as the result of a mutation, these DNA repair complexes do not form after DNA damage. Therefore, cells that are missing BRCA1 or BRCA2 are hypersensitive to agents that damage DNA, such as UV light and various chemical carcinogens. Women with an abnormal BRCA1 or BRCA2 gene, for example, have an extremely high risk of developing breast cancer and a high risk of developing ovarian cancer. As a result, many women who have a family history of breast and ovarian cancers undergo genetic testing. If they test positive for either gene mutation, they may opt to undergo a preventative mastectomy. Actress Angelina Jolie, who tested positive for a BRCA1 mutation and opted to undergo a preventative double mastectomy, recently brought this issue to the spotlight. While the publicity she generated was controversial, Jolie reduced her risk for developing breast cancer from 87 to less than five percent.
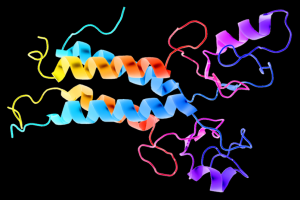
This is a 3D schematic representation (also known as a ribbon diagram) of the BRCA1 protein. BRCA1 is important for repairing damaged DNA, and BRCA1 mutations result in an increased risk for breast and ovarian cancer. Image credit: Wikimedia Commons
In addition to being crucial for DNA repair, BRCA1 and BRCA2 are also involved in cell cycle control. The Retinoblastoma protein, which detects damaged DNA, acts as the brake in cell cycle progression. Once they detect damaged DNA, BRCA1 and BRCA2 are crucial for the activation of these cell cycle checkpoints. BRCA1 also activates P53, a key protein involved in activating apoptosis in response to irreparable DNA damage. These essential roles for BRCA1 and BRCA2 proteins highlight their role in DNA repair, underscoring their function as molecular caretakers in our genome surveillance system.
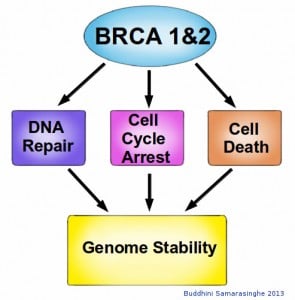
BRCA1 and BRCA2 proteins have many roles within a cell. Although their primary role is in DNA repair, they are also involved in halting the cell cycle upon detection of damaged DNA, utilizing cell cycle checkpoints. BRCA proteins are also able to activate cell death if the CNA damage is too extensive for repair. Thus BRCA proteins are essential for maintaining the stability of our genome, and without them we are at increased risk for cancer. Image credit: Buddhini Samarasinghe
Stacking The Deck
When our genome surveillance system is compromised, the rate of mutations increases (similar to how anarchy breaks loose when local law enforcement is indisposed). The effect of a mutation is still random, but cancer cells can stack the deck in their favor when the genome surveillance system is compromised, ensuring that future mutations occur more frequently. When cancer cells evolve, there is selection for mutations that can overcome the ten anticancer defense mechanisms represented by the Hallmarks of Cancer. Mutations in Ras allow cancer cells to remain independent from growth factors. Mutations in Retinoblastoma remove the brakes, allowing for uncontrolled cell division. P53 mutations allow cells to escape cell death. All of these mutations are produced initially by direct DNA damage, and secondarily as a result of mutations in genes that increase the probability of more mutations (i.e. mutations in DNA repair). Thus, as cancer cells continue to evolve and cells with specific mutations are selected, cells that carry mutations in genes that normally function in maintaining the stability of the genome are also selected. Genomes vary widely between different tumor types, but nearly all of them have DNA repair defects. Thus, genome instability and mutation are essential to tumor progression.
Reprinted with permission of Buddhini Samarasinghe and Scientific American, November 26, 2013
8. Tumor-Promoting Inflammation
We consider the immune system as our friend; it protects us by fighting infections while keeping us healthy. But there is a darker side to the immune system. Often, when it comes to cancer, we find that the immune system can turn traitor and actually promote cancer development.
The presence of white blood cells in tumors has been noted for many decades and provides the first clue that inflammation is linked to cancer. Yet it is only within the last few years that we have obtained clear evidence that inflammation plays a critical role in cancer development, and we are just beginning to understand the molecular mechanisms of how this happens. Indeed, chronic infections, obesity, smoking, alcohol consumption, environmental pollutants and high fat diets are now recognized as major risk factors for most common types of cancer; and, importantly, all these risk factors are linked to cancer through inflammation.
So what exactly is inflammation? It is how tissues and cells respond to injury. We are all familiar with the symptoms of acute inflammation: pain, heat, swelling and redness. The acute inflammatory response is normally localized and is protective. However, if the inflammation-causing agent persists for a prolonged period of time, the body's response to it becomes a chronic inflammation. This can happen because of infection, environmental pollutants, persistent activation of inflammatory proteins, or autoimmune reactions. Most importantly, chronic inflammation increases cancer risk. A slow smoldering fire in this case does far more damage than a flash fire.
The Wound That Doesn't Heal
What happens at the cellular level during inflammation? It is an extremely complex cascade of signaling molecules and cells that work together at the site of injury. Various cell types send out signaling molecules to attract different types of white blood cells. Of these, neutrophils are usually the first responders, and these cells kill microbes by engulfing them and releasing chemicals collectively known as reactive oxygen species. ROS kill pathogens and nearby healthy cells alike-they are messy. If an inflammation persists and becomes chronic, there is a shift in the type of white blood cell found nearby, the primary type being macrophages. Chronic inflammation causes tissue damage and often results in the repair of this damaged tissue by replacement with fibrous connective tissue.
An extremely graphic yet apt way of describing a tumor is as "a wound that doesn't heal." Indeed, there are many similarities between a cancerous tumor and the process of wound healing. Both involve the growth, survival and migration of cells; both require the growth of new blood vessels. Importantly, all these processes are controlled by growth factors and signaling molecules. Just as the immune cells gather near a site of injury to secrete growth factors to begin tissue repair, tumors can also surround themselves with immune cells that secrete these same growth factors to promote their uncontrolled cell growth.
NF-κB: The First Violin
What orchestrates the inflammatory response? A protein known as NFκB (pronounced NF-kappa-B) has been described as "playing the first violin, if it is not the conductor of the inflammatory response." Since NFκB is such an important first responder to inflammation it is already present in cells in an inactive state and does not require any protein synthesis to become activated. NFκB is held in an inactive state by a protein known as IκBα, which binds to NFκB and prevents it from moving into the nucleus of the cell. IκBα essentially behaves like a goalkeeper. So to activate the NFκB pathway, the goalkeeper must be neutralized. This is the role of an enzyme known as IKK (IκB kinase). Remember, kinases are enzymes that add phosphate groups to proteins. By adding a phosphate group to IκBα the goalkeeper, IKK 'tags' it for degradation; the goalkeeper is neutralized. NFκB is now free to move into the nucleus of the cell and activate a wide range of genes that promote cell growth, survival, migration and the formation of new blood vessels. In the context of cancer, the activation of NFκB in immune cells induces the production of molecules that activate NFκB in cancer cells; these in turn induce molecules that attract more immune cells into the tumor. It is a feed-forward loop that results in a frenzy of uncontrolled growth, with the immune cells actively enhancing the Hallmark capabilities of the cancer cells.
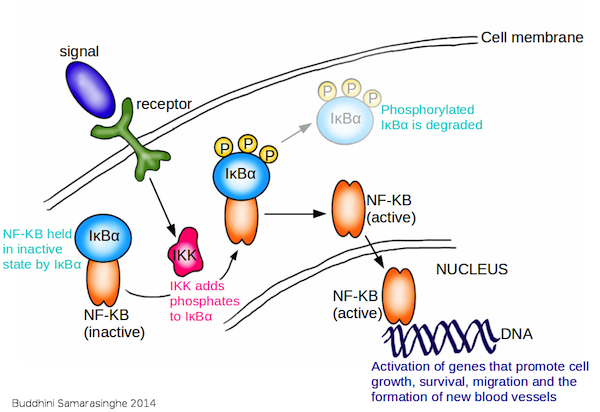
The activation of the NFκB pathway is a key event in the cellular inflammatory response. Under normal circumstances, NFκB is held in an inactive state by IκBα. The binding of a signal molecule to a specific receptor activates the IKK protein. Activated IKK adds phosphate molecules to IκBα, tagging it for degradation. NFκB is now active, and moves into the nucleus to activate genes that promote cell growth, survival, migration and the formation of new blood vessels. Image by Buddhini Samarasinghe.
TAMs Are Bad News
Although various types of immune cells are involved in this process, with respect to supporting tumors, the main culprits are known as Tumor Associated Macrophages (TAMs). Patients with tumors that have been infiltrated by TAMs have a much poorer prognosis than those that do not. These TAMs are found in most malignant tumors, and in some instances can comprise up to 50 percent of the cell tumor mass. TAMs can support a growing tumor by assisting the cancer cells to bypass some of the anti-cancer defense mechanisms that are represented by the Hallmarks. There are four main mechanisms by which TAMs help a tumor develop and grow.
First, a tumor is addicted to a constant supply of growth factors, which it uses to drive its unchecked growth as described in Hallmark 1. Although the tumor cells themselves often carry mutations that allow them independence from this limitation, TAMs also provide the necessary growth factors. TAMs have been shown to produce growth factors such as Epidermal Growth Factor (EGF), Fibroblast Growth Factor (FGF), and other small molecules known as cytokines, primarily IL-6 and TNF.
Second, as described in Hallmark 5, a growing tumor needs to establish a blood supply through a process known as angiogenesis. Angiogenesis is primarily regulated through the angiogenic growth factor Vascular Endothelial Growth Factor (VEGF) and Platelet Derived Growth Factor (PDGF). TAMs have been shown to secrete both VEGF and PDGF into the tumor microenvironment in order to promote angiogenesis. The uneven distribution of blood vessels and regions of oxygen starvation (termed hypoxia) are characteristics of advanced tumors, and TAMs often preferentially accumulate in these regions, activating angiogenesis through the hypoxia response pathway as described in Hallmark 5. Intriguingly, VEGF has also been shown to attract TAMs to a tumor, demonstrating yet another feed-forward loop where TAMs surrounding a tumor secrete VEGF which in turn attracts more TAMs to that tumor.
Third, as described in Hallmark 6, the spread of cancer cells from the site of the primary tumor into distant metastases requires the degradation of the Extra Cellular Matrix (ECM). This is aided by enzymes known as matrix-degrading enzymes, notably matrix metalloproteases and again TAMs have been shown to secrete these enzymes to aid the invasion and spread of cancer cells to distant sites within the body.
Finally, TAMs can suppress the adaptive immune system, so that any cancer-killing activity of the immune system is effectively neutralized. TAMs do this by producing immunosuppressive molecules, thus allowing the tumor to evade the immune system. TAMs can also release cytokines that preferentially attract other immune cells that have no cell-killing activity. It is the equivalent of recruiting law enforcement officers carrying toy pistols-the tumor is unaffected and indeed thrives thanks to being unimpeded by the tumor-killing aspects of the immune system. This process will be discussed later in Hallmark 10.
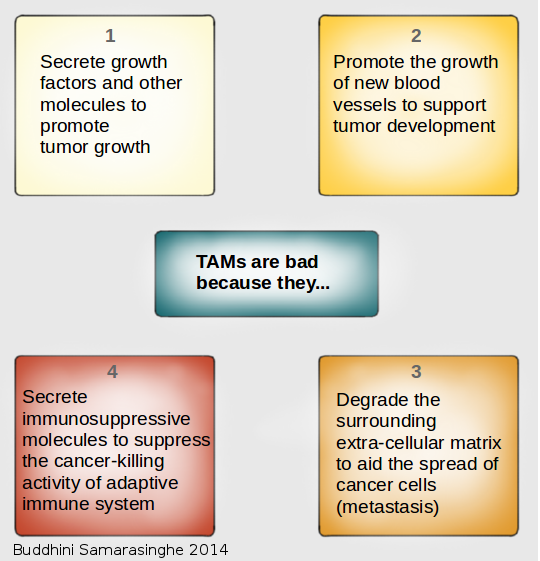
Tumor associated macrophages (TAMS) support tumors through four different mechanisms. Image by Buddhini Samarasinghe.
Feed-forward Loop of TAM Recruitment
How are ordinary macrophages recruited and corrupted to become TAMs? Intriguingly, a developing tumor does not recruit existing macrophages into becoming TAMs. Instead, tumor cells produce many small molecules known as chemokines and cytokines during the early development of the tumor. These molecules 'convert' precursor macrophages (known as monocytes) into TAMs. As the tumor progresses, areas of low oxygen develop; this results in the activation of the hypoxia response pathway (described in Hallmark 5) which liberates VEGF and other factors and also includes the activation of NFκB. This in turn attracts even more TAMs to areas of low oxygen.
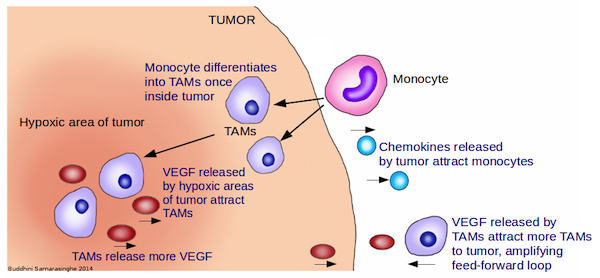
Tumors and their TAMS. Tumors secrete signalling molecules known as chemokines to attract circulating monocytes, a type of white blood cell. Once in the tumor, the monocytes differentiate into tumor associated macrophages (TAMS). oxygen-starved (hypoxic) areas of the tumor secrete VEGF, which attracts these TAMS. TAMS can also secrete VEGF, which in turn attract more TAMS to the tumor. Image by Buddhini Samarasinghe.
It seems counterintuitive to consider the existence of both tumor-promoting and tumor-killing immune cells. Upon considering the complex and diverse roles of the immune system it is easier to understand. The immune system specifically detects and targets infections through the adaptive immune system (the specific branch utilized by vaccines) which is supported by cells of the innate immune system (the non-specific branch). In turn, the innate immune system is involved in wound healing and clearing up dead cells. Specialized immune cells are responsible for each of these varied functions. The immune cells specializing in wound cleaning and clearing up dead cells are recruited and subverted by tumor cells to support tumor growth and progression. Shifting the balance from tumor promoting to tumor killing immune involvement therefore represents an attractive and powerful possibility for future therapy.
Reprinted with permission of Buddhini Samarasinghe and Scientific American, April 18, 2014
9. Reprogramming Energy Metabolism
Uncontrolled growth defines cancer. Growth requires a cancer's cells to replicate all of their cellular components; their DNA, RNA, proteins and lipids must all be doubled in order to divide into daughter cells. Of course, this process requires energy. Cancer cells must adjust their metabolism accordingly to enable this frenzied growth.
Respiration 101
Cells require energy to absorb nutrients, to react to changes in their surroundings, to maintain their internal environment, grow and replicate. Energy is obtained from breaking down nutrients through a process of metabolic reactions known as respiration. This energy is stored in small molecules known as adenosine triphosphate, or ATP. When cells need energy, they use respiration to construct molecules of ATP; they then break down the ATP to fuel their metabolic reactions. Respiration comes in two forms. Normal cells, under normal conditions, undergo aerobic respiration, which is a metabolic pathway that requires oxygen. Cells break down glucose into pyruvate, to eventually form ATP, while releasing carbon dioxide as a waste product. The oxygen needed is obtained from the air we breathe, diffuses into our blood and is then transported across all our tissues and organs. When there isn't enough oxygen, cells switch to a different type of respiration called anaerobic respiration. During anaerobic respiration, cells break down glucose into pyruvate and construct ATP, but produce lactic acid instead of carbon dioxide. Aerobic respiration produces far more ATP molecules, 32 per molecule of glucose, than anaerobic respiration, which produces a mere two.
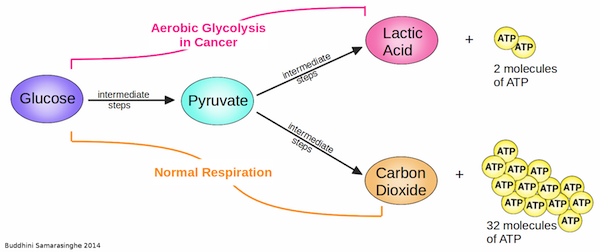
In cancer cells undergoing aerobic respiration (pink pathway), glucose gets broken down into pyruvate and then lactic acid, producing only two molecules of ATP. In cells undergoing normal respiration (orange pathway), glucose gets completely broken down into pyruvate, which is further processed into carbon dioxide, producing 32 molecules of ATP. Image credit: Buddhini Samarasinghe
Aerobic Glycolysis
Glycolysis is the process in which cells break glucose down into pyruvate, the first step in constructing the energy transfer molecule ATP. One of the main observations of the metabolism of cancer cells is that they display an enhanced rate of glycolysis during periods of fast growth. This phenomenon is known as the Warburg Effect, after its discoverer Otto Warburg, and is also known (somewhat confusingly) as aerobic glycolysis. Cancer cells consume more than 20 times as much glucose compared to normal cells, but secrete lactic acid instead of breaking it down completely into carbon dioxide. Why do cancer cells choose this inefficient metabolic pathway when they could obtain 16 times as much ATP per molecule of glucose by opting for normal respiration?
The answer is twofold. First, although cancer cells produce far less ATP per molecule of glucose, they produce it much faster. Cancer cells produce ATP almost a hundred times faster than normal cells. It is essentially a cost-benefit calculation, where the benefits of speedy ATP production outweigh the costs associated with inefficient glucose breakdown. Second, it is not just about ATP production. Cancer cells undergoing aerobic glycolysis also produce many intermediate biosynthetic precursors. These molecules are used as building blocks for the production of proteins, lipids and DNA required by the rapidly dividing cells.
Greedy for Glucose
How do cancer cells satisfy their voracious appetite for glucose? Glucose typically enters cells through protein channels known as glucose transporters, which act as gateways through the surface of the cell membrane, selectively allowing glucose molecules to enter the cell. Cancer cells actively produce more glucose transporters on their cell surface membranes, so more glucose is brought inside the cell. Once inside the cell, the glucose is broken down by aerobic glycolysis into lactic acid, in order to speedily produce ATP and metabolic precursors through various metabolic pathways. These pathways are tightly controlled, requiring specific enzymes for processing the molecules from each step to the next. Cancer cells are addicted to these metabolic precursors; the enzymes that control these pathways are often over-expressed or mutated in cancer cells. This addiction is exploited in chemotherapy strategies. For example, the drugs 5-fluorouracil, methotrexate and pemetrexed inhibit the biosynthesis of DNA precursor molecules. The high glucose consumption of cancer cells is also exploited when imaging cancer. Positron emission tomography (PET) combined with computer tomography (PET/CT) is used to detect the absorption of the glucose analogue fluorodeoxyglucose (FDG) by tumors, and has a better than 90 percent sensitivity and specificity for the detection of metastases of most cancers.
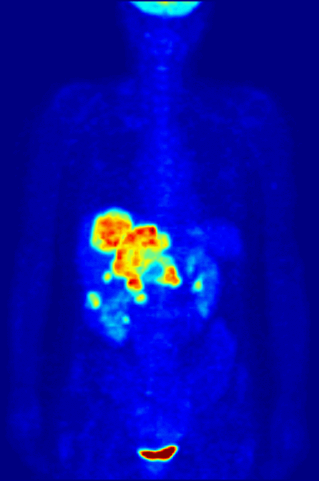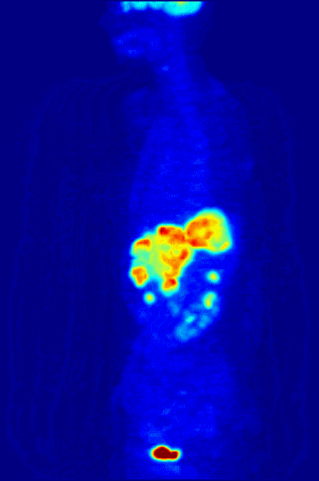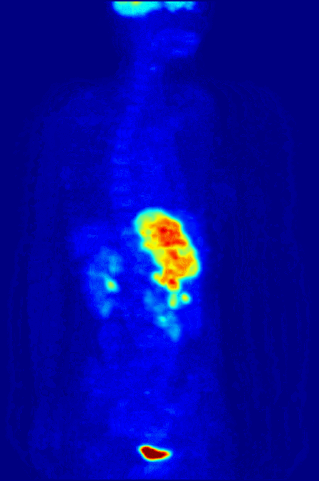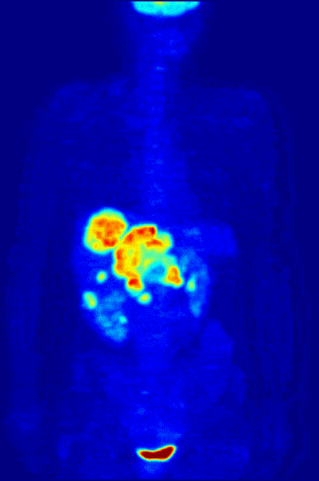
Cancer cells consume more glucose than normal cells. This is exploited when imaging cancer. Positron emission tomography (PET) combined with computer tomography (PET/CT) is used to detect the absorption of the glucose analogue fluorodeoxyglucose (FDG) by tumours. In this image, besides the normal accumulation of the FDG molecule in the heart, bladder, kidneys, and brain, liver metastases of a colorectal tumor are visible in the abdominal region. Image credit: Jens Maus, Wikimedia Commons
What is the utility of this alternative metabolic pathway to healthy cells? The ability to grow and divide rapidly is useful in the context of wound healing and immune responses. When an immune response is required, immune cells massively increase their glucose uptake, switch from metabolizing glucose through normal respiration to aerobic glycolysis, and switch on the multitude of enzymes that control the biosynthesis of proteins, lipids and DNA. Therefore, there is a strong evolutionary basis for rapid cell division and faster growth despite the inefficient use of glucose in the process. Cancer cells hijack this metabolic switch in order to fuel their own uncontrolled growth.
The Mechanisms of a Metabolic Switch
We therefore know why cancer cells opt to switch from normal respiration to aerobic glycolysis. The next question is how they do this. This is an active area of research, and we are still deciphering the exact mechanisms of this metabolic switch. In a previous article, I explained how tumors often develop regions of low oxygen. This activates the hypoxia stress response, mediated by the hypoxia inducible factor (HIF). However, over recent years, it has become clear that HIF activity is not merely a response to low oxygen levels. HIF can be activated in response to a variety of triggers, such as radiation induced DNA damage, signaling from other proteins, growth factors and the presence of pyruvate. Once activated, HIF can go on to activate genes that support aerobic glycolysis and repress genes involved in normal respiration. HIF clearly plays a central regulatory role in switching the metabolism of cancer cells from normal respiration to aerobic glycolysis. But what are the other signaling pathways that support this metabolic switch? One of the most important pathways is controlled by an enzyme known as Phosphoinositide 3 Kinase (often shortened to PI3K). PI3K can be activated by a variety of survival signals from outside the cell via receptors on the cell surface membrane. Once activated, the PI3K pathway can mediate cell growth and survival, inhibit apoptosis, activate angiogenesis and, notably, activate aerobic glycolysis. In other words, activation of the PI3K pathway supports cancer cells. PI3K activates another protein known as AKT, which then activates the protein mTOR. Once activated, mTOR can also activate HIF, further supporting HIF's efforts to switch cancer cell metabolism from normal respiration to aerobic glycolysis. PI3K signaling is negatively regulated by another protein known as PTEN. It is therefore unsurprising that cancers often have mutations that inactivate PTEN, thus preventing it from inactivating PI3K signaling.
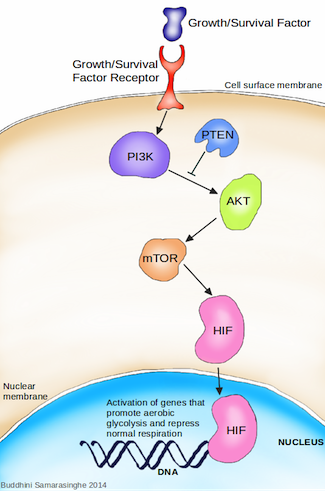
Switching metabolism in cancer. Growth or survival factor signaling activates the PI3K signaling pathway. activated PI3K activates AKT, which then activates MTOR, which then activates HIF. HIF moves into the nucleus of the cell and activates genes that promote aerobic glycolysis while repressing normal metabolism. Activated PI3K can be attenuated by tumour suppressor protein PTEN. Image credit: Buddhini Samarasinghe
It is increasingly important to understand that cancer progression is not so much a signaling pathway as it is a signaling web. Throughout this series, I have described normal cellular processes that serve to check uncontrolled growth and division. Many of the signaling pathways involved in these processes can cross-talk with each other: the components of one pathway can regulate another. For example, I discussed how the protein P53 is known as the Guardian of the Genome, as it is responsible for detecting and mitigating DNA damage. Another recently discovered function of P53 is in promoting normal respiration while dampening aerobic glycolysis. Given that P53 is the most commonly mutated protein in all cancers, it is unsurprising that the loss of normal P53 function disrupts this balance and pushes the cell towards aerobic glycolysis. The cellular pathways described in the Hallmarks of Cancer are not isolated; when mutated, they feed into each other, amplifying their effects and driving cells towards the uncontrolled growth that results in cancer.
It is also important to understand that cancer cells do not function in isolation. Tumors are complex tissues, made up not only of cancer cells, but also blood vessels, immune cells and other bystanders. These healthy cells are co-opted and subverted to perform tasks that support cancer progression. For example, the lactic acid secreted by cancer cells can induce tumor associated macrophages to create new blood vessels for the growing tumor via the HIF pathway. The cellular pathways described in the Hallmarks of Cancer are not only interconnected; the body's own cells, when trapped inside the tumor microenvironment, engage in complementary metabolic pathways, supporting and encouraging cancer cell survival and growth.
Reprinted with permission of Buddhini Samarasinghe and Scientific American, October 8, 2014